Adult Orbital Xanthogranulomatous Disease
Updated July 2024
Brittany A. Simmons, MD; Keith D. Carter, MD, FACS; Elizabeth A. Bradley, MD; and Erin M. Shriver, MD, FACS
Establishing the diagnosis
Adult xanthogranulomatous disease of the ocular adnexa and orbit comprises a heterogenous group of infiltrative non-Langerhans cell, histiocytic inflammatory syndromes that may be isolated to the orbit or associated with systemic disease. From most to least frequent, adult xanthogranulomatous disease is classified as necrobiotic xanthogranuloma (NBX), Erdheim-Chester disease (ECD), adult-onset asthma with periocular xanthogranuloma (AAPOX), and adult-onset xanthogranuloma (AOX). While histologically similar, each disease exhibits distinct manifestations and prognoses, and the collective rarity of this disease entity has made effective management difficult. Of note, juvenile xanthogranuloma is a separate non-Langherans cell histiocytic entity that is a focal subcutaneous, self-limited, and corticosteroid sensitive disease of childhood, and is beyond the scope of this review.
Etiology
- The causative etiology of xanthogranulomatous disease is unknown.
- May be immunologically mediated with possible histiocyte-stimulating insult.
Epidemiology
- Approximately 200 total cases of adult orbital xanthogranulomatous disease have been published in the English language literature to date, with fewer than 50 cases for some disease subsets.
- Adult xanthogranulomatous disease is diagnosed in middle age (average 40s to 50s years of age, range 17-85 years).
- There is no sex predilection for AOX. NBX may have a female predominance. AAPOX and ECD are 2-3 times more common in males.
- NBX and AAPOX are associated with immune dysfunction, including asthma, lymphadenopathy, paraproteinemia, or other lymphoproliferative disorders.
Clinical features
- AOX, AAPOX, and NBX are associated with more preseptal and anterior orbital disease compared to the almost uniform, diffusely posterior and/or intraconal disease seen with ECD.
- 42-95% of patients have eyelid skin lesions. When present, these lesions appear as yellowish, subcutaneous plaques in the periorbital region. This is the only manifestation of disease in AOX.
- AAPOX is the combination of periorbital lesions with adult-onset asthma, the latter of which may be diagnosed by pulmonary function tests showing reversible airway obstruction. Patients may also have chronic rhinosinusitis and nasal polyps.
- NBX is more aggressive, with indurated yellow papule or plaque-like skin lesions that ulcerate or fibrose. There is eye involvement in 50% of cases with keratitis, uveitis, or proptosis. Skin lesions may arise elsewhere on the body, and may rarely involve multiple systems including the GI tract, heart, lung, brain, and muscle. Nearly all cases of NBX have some form of immune dysfunction with subsequent paraproteinemia, monoclonal gammopathy of undetermined significance, multiple myeloma, leukemia, and other lymphoproliferative disorders. There is a higher incidence of elevated body mass index and low high-density-lipoprotein-cholesterol levels in these patients.
- ECD more commonly presents as a posterior, often bilateral, orbital mass. Signs and symptoms include progressive, painless vision loss, ophthalmoplegia, and proptosis. The appearance of orbital congestion may be present, as well as ocular surface irritation with keratopathy in the setting of marked proptosis. Internal organ dysfunction – including pericardial or pleural effusion, mediastinal or retroperitoneal infiltrates, brain or dural masses – is almost always seen in ECD. Systemic findings relate to extra-orbital areas of disease, such as bone pain from sclerosis of the long bones or excessive thirst and urination with diabetes insipidus from pituitary involvement. ECD is associated with a higher rate of myeloid malignancy.
Establishing the diagnosis
Diagnosis entails biopsy with classic histopathologic findings. Because some histopathologic findings are common to all forms of disease, further classification relies on systemic manifestations of illness and, in the case of ECD, molecular studies.
- Non-Langherhans lipid laden foamy histiocytes (“xanthoma cells”) and Touton multinucleated giant cells, and variable lymphocytic infiltrate, fibrosis, and possibly necrosis. Xanthoma cells are positive for CD68, CD163, and factor XIIIa, and rarely positive for S100 with a negative factor XIIIa (such as in juvenile xanthogranuloma).
- Laboratory evaluations may show leukocytosis, eosinophilia, elevated immunoglobulins (including IgG), elevated rheumatoid factor, and decreased complement factor C4.
- In the orbit, neuroimaging may show homogenous, enhancing tissue with indistinct borders in the preseptal or postseptal regions that is often bilateral with concurrent changes in the muscles (such as enlargement) and even the optic nerve (with compression or thinning). Unilateral disease is also possible. Computed tomography (CT) shows lesions typically isointense to muscle. Magnetic resonance imaging (MRI) lesions are iso- or slightly hyperintense (T1) or isointense (T2) to muscle. Lesions can be infiltrative with bony destruction (ECD), and may involve the lacrimal gland.
Disease classification
Xanthogranulomatous disease is classified based on histopathology and systemic manifestations.
- AOX is strictly periocular or periobital in nature with no systemic lesions.
- AAPOX displays periocular xanthogranulomas associated with asthma. Associated with rhinosinusitis, nasal polyps, increased IgE and IgG levels, and lymphadenopathy.
- NBX shows lesions of the eyelid skin and anterior orbit, and possible similar cutaneous lesions throughout the body that ulcerate and fibrose. 50% have ocular involvement with keratitis, uveitis, and proptosis from orbital lesions. Systemic associations include immune dysfunction, paraproteinemia, multiple myeloma, and other lymphoproliferative disorders.
- ECD presents as an orbital mass, most commonly bilateral, diffuse and intraconal. Lesions have dense, progressive fibrosclerosis of the orbit as well as involved organs. Can affect the mediastinum, perinephric and retroperitoneal space, pericardium, pleura, brain, and bone. ECD is a systemic clonal disorder displaying a BRAF V600E mutation in the majority of cases, and, as such, is one of the only xanthogranulomatous diseases to employ complementary molecular studies for diagnosis.
Risk factors
- No modifiable risk factors for xanthogranulomatous disease identified at this time.
- Association between some xanthogranulomatous diseases and paraproteinemia or lymphoproliferative disorders, but unclear if these are triggers versus concurrent manifestations of the disease.
- A mutation in BRAF V600E may predispose to ECD; however, this is a somatic mutation and not a heritable, germline mutation.
Differential diagnosis
- The differential diagnosis for adult orbital xanthogranulomatous disease includes xanthelasma; granulomatous diseases like sarcoidosis, granulomatosis with polyangiitis, and foreign body reaction; lymphoproliferative disorders; amyloidosis; Rosai-Dorfman disease; Kimura disease; idiopathic orbital inflammation; thyroid eye disease; IgG4 disease; and Langerhans cell histiocytosis.
Patient management: treatment and follow-up
Workup
Evaluation of the patient with adult xanthogranulomatous disease requires exploration of orbital involvement, hematologic disease, and infiltration of peripheral organs. Workup is then dictated by the type of xanthogranulomatous disease.
- Complete ophthalmic exam with attention to ocular or orbital inflammation, optic nerve dysfunction, dysmotility, and proptosis. Baseline ophthalmic testing (such as optic nerve optical coherence tomography and formal visual field testing) if there is an orbital mass or concerning orbital findings on exam.
- Neuroimaging (CT or MRI) for an orbital mass. Systemic imaging with CT of the chest, abdomen, and pelvis.
- Biopsy with flow cytometry and histopathologic analysis.
- After diagnosis, systemic evaluation in conjunction with the patient’s primary care physician. Assess for asthma, other skin lesions, or positive findings on review of systems that may point to other systemic lesions.
- Labs including a complete blood count with differential, comprehensive metabolic panel, serum protein electrophoresis, and urine studies may be obtained.
- Specialty-specific imaging and additional studies such as bone marrow biopsy may be indicated pending the results of these initial evaluations. For diagnoses of NBX or ECD, referral to a hematologist-oncologist is warranted.
Treatment
Treatment is variable and is dictated by the location and extent of disease.
- Ocular inflammation and keratopathy are managed with topical anti-inflammatory medications and lubrication. Some patients may require temporizing measures to protect the ocular surface, such as a tarsorrhaphy.
However, these therapies are aimed at treating the manifestations of disease and do not target the underlying disease process. Due to the overall rarity of these disorders, there have been no randomized control trials studying treatment, and systemic treatment recommendations are derived from published case reports.
- For suspected AOX, biopsy and surgical excision of lesions may be sufficient.
- While isolated reports exist of sufficient improvement after surgical debulking of orbital lesions, in most cases surgery is often limited to biopsy. Excisional surgery is considered controversial due to possible stimulation of disease with risk of relapse, as well as difficulty in resecting these lesions.
- One publication reported spontaneous regression of orbital AOX over 10 years of observation. While some patients can be monitored without systemic treatment, spontaneous improvement is not a known feature of these disorders and patients often have problematic symptoms such as diplopia, proptosis, or optic neuropathy that require some form of medical management.
- Intralesional steroid injection for periorbital xanthogranulomas may have some benefit, though this generally does not completely resolve disease.
- Unresectable and systemic disease has traditionally been managed with long-term low dose systemic steroids, though the disease may flare as steroids are tapered.
- A better response may be obtained with antimetabolites and immunomodulators like methotrexate, azathioprine, cyclophosphamide, cyclosporin, and rituximab. Chemotherapy regimens may offer only transient relief of symptoms.
- NBX may respond well to treatment regimens including IVIG, antimalarials, intralesional steroid injection, extracorporeal photophoresis, and autologous hematopoietic progenitor cell transplants.
- Interferon alfa is considered by some to be a first-line approach to ECD treatment.
- BRAF inhibitors like vemurafenib may improve outcomes in ECD.
- Radiation has variable results, with some reports showing good efficacy while others report no benefit or exacerbation of xanthogranulomatous disease.
Follow up
Follow up should be regular and long-term. However, given the variability of disease and response to treatment, there is no prescribed follow up schedule.
- Follow up relies more on clinical findings than serial imaging.
- Increased incidence of hematologic malignancy requires long-term follow up with special attention paid to a thorough review of systems and regular blood work.
Prognosis
Prognosis depends on the variant of disease.
- AOX, considered a benign cutaneous disease, has an excellent prognosis.
- AAPOX is also considered to have a favorable prognosis.
- NBX is a chronic, slowly progressive disease, but boasts a 90% survival at 15 years.
- CD has a survival rate of 70% at 5 years and 33% at 15 years.
Complications
Related to disease
Disease-related complications are directly correlated to areas of disease manifestation.
- NBX has ulcerative and fibrosing skin lesions that can lead to eyelid malposition. While the disease should be controlled systemically, surgical correction of eyelid malposition may be considered. Weigh the benefits of improved eyelid position and function with any risk of exacerbating the underlying disease.
- Patients with NBX almost invariably develop blood dyscrasias, and should be monitored and treated accordingly.
- ECD has the most significant systemic complications, including but not limited to long bone osteosclerosis with bone pain, neurologic dysfunction with exophthalmos, seizures, and diabetes insipidus, and constitutional symptoms of fever, weight loss, and fatigue, followed less frequently by dyspnea and/or pericardial effusion. Care is multidisciplinary.
Related to treatment
- High rate of disease recurrence after any surgical resection suggests that, rather than repeat removal of the lesion, systemic therapy should be undertaken.
- No standardized therapy recommendations. Numerous case reports and anecdotal treatment recommendations. For these reasons, it is impossible to recommend generic management of treatment complications.
- Oculoplastic surgeons, primary care providers, and hematologist-oncologists should work together to manage any disease- and treatment-specific complications.
Historical perspective
Since 1987, the classification of non-Langerhans cell histiocytosis includes numerous disorders in addition to those mentioned above, including juvenile xanthogranuloma, sinus histiocytosis with massive lymphadenopathy, and indeterminate cell histiocytosis to name but a few.
The presence of an increased number of IgG4+ cells in tissue samples with xanthogranulomatous disease, as well as a handful of patients diagnosed with both orbital xanthogranulomatous disease and IgG4-related disorders, has recently raised the question of a link between xanthogranulomatous disease and IgG4 disease. It is unclear if these diseases represent a continuum or are instead two separate entities with only some shared overlap in non-pathognomonic features.
The four entities discussed above are the current accepted classification; however, increasing investigation of these pathologies — including genetic studies and immuno-phenotypic signatures — may subsequently separate them into more disparate diseases.
Table: Summary of adult onset orbital xanthogranulomatous disease
Summary of adult onset orbital xanthogranulomatous disease
| Adult Onset Xanthogranuloma (AOX) |
Adult onset Asthma and Periocular Xanthogranuloma (AAPOX) |
Necrobiotic Xanthogranuloma (NBX) |
Erdheim-Chester Disease (ECD) |
|
|---|---|---|---|---|
| Gender predilection | None | F > M | M > F | M > F |
| Location of disease | Periorbital |
|
|
|
|
Systemic disease associations |
None |
|
|
None* |
| Histopathologic features** | Xanthomatous histiocytes, Touton giant cells, lymphocytic infiltrate | Lymphoid follicles with germinal centers | Necrobiosis with palisading histiocytes, cholesterol clefts | Fibrosis |
| Treatment |
|
|
|
|
| Prognosis | Excellent | Favorable |
Slowly progressive
|
Progressive
|
*Systemic disease can include site-specific sequelae of disease (such as liver enzyme abnormalities if liver disease in NBX, or hypertension from retroperitoneal fibrosis in ECD); for purposes of this table, the associations listed are commonly accepted disease entities associated with each subset of adult onset orbital xanthogranulomatous disease.
**Foamy histiocytes, Touton giant cells, and a lymphocytic infiltrate are features of all types. Additional features more commonly seen in each subtype of disease are listed here.
Cases
Case 1. Erdheim Chester disease
Figure 1. A 55-year-old Caucasian woman was referred for evaluation of new onset blurry vision and right eye pain. Her external and ophthalmic exams were within normal limits bilaterally.
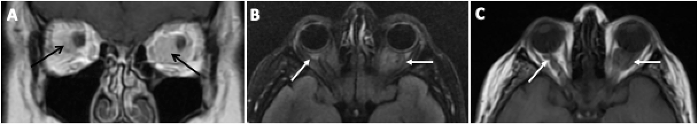
Figure 2. Neuroimaging was obtained. MRI shows bilateral orbital masses (arrows). A) Coronal T1 post-contrast shows enhancement of the masses that are homogenous; B) axial T2 FLAIR fat saturation illustrating the masses abutting or wrapping around the optic nerves; and C) axial T1 also demonstrating an intraconal location.
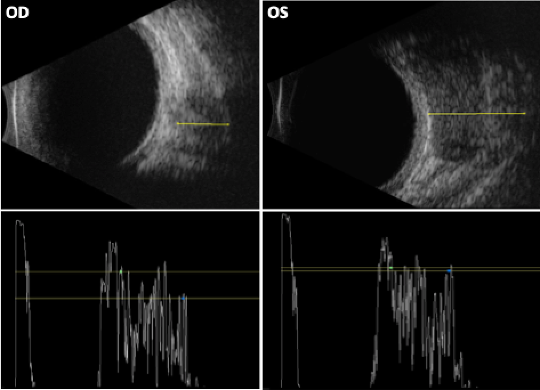
Figure 3. Orbital echography demonstrated bilateral mass lesions around the optic nerves with medium reflectivity and a heterogenous and irregular internal structure. There was possible vascularity within the lesions. The lesion on the right measured 6.69mm and the left was 12.91mm (denoted by the yellow lines, respectively). OD = right, OS = left.
The patient underwent biopsy of the left orbital mass.
Figure 4. Histopathology of the left orbital mass showed xanthogranulomatous disease with epithelioid histiocytes and xanthomatous macrophages, with inflammatory cells, lymphocytic aggregates, and fibrocollagenous septae. These cells were positive for CD163 and negative for CD1a, with infrequent IgG cells and IgG4 positivity in 25-30% of plasma cells.
Flow cytometry showed no immunophenotypic evidence of a non-Hodgkin’s lymphoma. Molecular testing revealed a BRAF mutation suggestive of Erdheim-Chester disease.
The patient established care with an oncologist, and initial workup with positive emission tomography (PET) CT showed FDG uptake in the intraconal left orbit and no other abnormalities. Bone marrow biopsy showed no evidence of myelodysplasia or infiltrative histiocytic process. Subsequently, the patient developed urinary symptoms with frequency and dysuria in the setting of right-sided hydronephrosis associated with inflammatory changes. It is unclear at this time if this is related to her ECD.
Case 2: Erdheim Chester disease
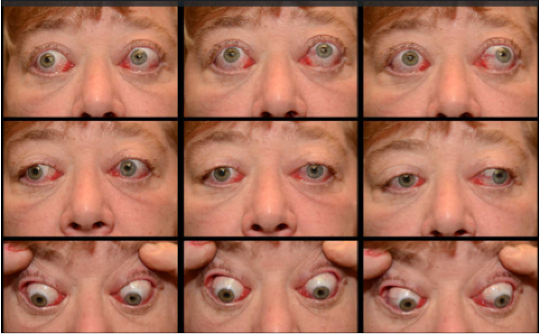
Figure 5. A 53-year-old Caucasian woman was referred for evaluation of bilateral proptosis and ocular irritation for 6 months. On presentation, her exam was notable for bilateral exophthalmos (Hertel exophthalmometry 26mm right, 24mm left, at a base of 106). Motility was restricted in all fields of gaze bilaterally, and she displayed signs of orbital congestion including periorbital edema, ocular surface injection, and caruncle swelling.
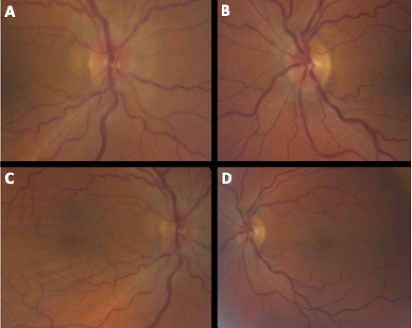
Figure 6. Her ocular exam showed decreased visual acuity of 20/250 (Snellen) on the right (in the setting of amblyopia of unknown degree) and 20/20 on the left. The posterior pole showed Frisen Grade 1 papilledema on the right (A) and early Frisen grade 1 papilledema on the left (B), with prominent choroidal folds throughout the macula on the right (C) and venous congestion on the right (C) as well as the left (D).
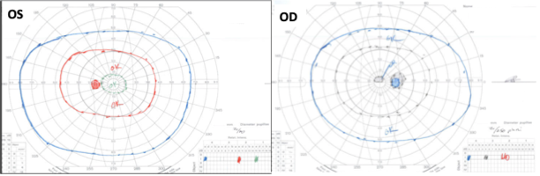
Figure 7. Diagnostic testing revealed Goldmann visual field testing with general depression with a small central scotoma on the right with a preserved visual field on the left (OS). Critical flicker fusion was moderately to severely decreased on the right and mildly decreased on the left.
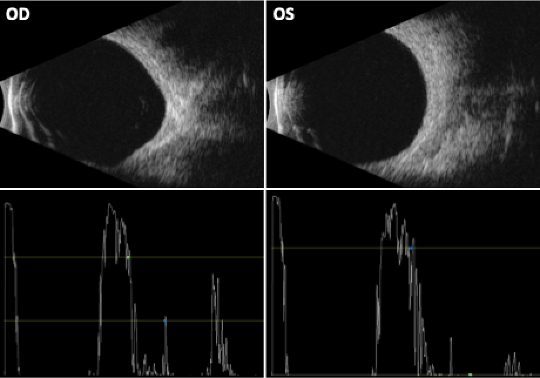
Figure 8. Orbital echography demonstrated globe deformation on the right (OD). Extraocular rectus muscles were enlarged on the right (3 of 4, excluding the right inferior rectus) and on the left (OS)(4 of 4). There was no distinct mass lesion, posterior scleritis, or signs of myositis detected.
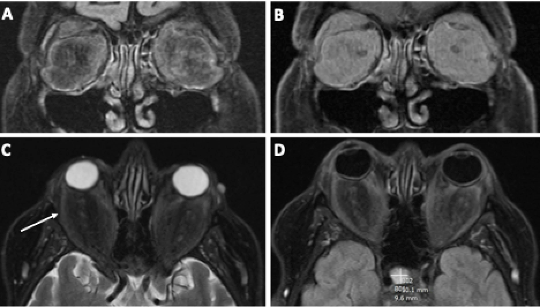
Figure 9. MRI of the brain and orbits showed bilateral contrast-enhancing retrobulbar soft tissue expansion. A) Coronal T2 fluid-attenuated inversion recovery (FLAIR) and B) coronal T1 fat saturation post-contrast showing hyperintense and enhancing retro-orbital soft tissue expansion, C) axial T2 fat saturation with stretched and atrophic-appearing extraocular muscles (arrow), and D) axial T2 FLAIR illustrating intraconal tissue expansion and a lesion in the pituitary (plus sign).
Laboratory workup including an array of infectious and autoimmune etiologies was unrevealing. The patient underwent biopsy of the right orbital mass.
Figure 10. Histopathology of the right orbital mass showed lacrimal gland with mild chronic inflammation and adjacent orbital tissue with a histiocytic infiltrate characterized by large foamy histiocytes. These cells were positive for CD163 and negative for CD1a. Molecular testing was positive for BRAF V600E mutation.
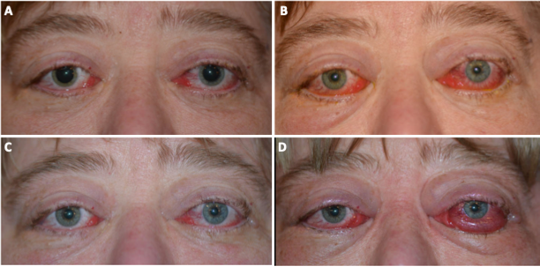
Figure 11. Clinical photos taken over the patient’s disease course show A) initial presentation with bilateral caruncle and ocular surface inflammation. Systemic steroid therapy was initiated. When she was unable to taper off steroids and showed worsening inflammation (B), rheumatology added mycophenolate mofetil. While the patient initially improved with mycophenolate (C), she acutely worsened after 1 month and subsequently completed low dose orbital radiation with radiation oncology (20 Gray in 10 fractions). Despite an initial improvement, she again worsened (D) with massive chemosis on the left necessitating a temporary tarsorrhaphy.
Due to her recalcitrant disease, the patient sought a second opinion. Four weeks later, she had a telemedicine visit with the second institution due to COVID-19. During that visit, it was noted that visual acuity and color vision on the left had decreased. She was started on high dose intravenous steroids, which she received locally. Despite this, 4 days later her vision precipitously declined to bilateral no light perception. She did not seek immediate evaluation, and when this came to the attention of her providers 1 day later, she underwent same day emergent bilateral orbital decompression. Unfortunately, her vision did not recover. Over the next 2 weeks, she developed massive chemosis necessitating a temporary tarsorrhaphy on the right. Concurrently, PET CT suggested cardiac involvement and she was also diagnosed with hydronephrosis and retroperitoneal fibrosis. The patient was started on the BRAF inhibitor vemurafenib as well as the MEK inhibitor cobimetinib with mixed response. While visual acuity remained no light perception, the orbital disease improved clinically and radiographically. Unfortunately, systemically the patient developed new foci of FDG uptake in a periportal lymph node, spleen, pelvic soft tissues, and pelvic bones. The patient also developed thrombocytosis. Re-staging workup is currently underway.
References and additional resources
- Arnaud L, Gorochov G, Charlotte F, et al. Systemic perturbation of cytokine and chemokine networks in Erdheim-Chester disease: a single-center series of 37 patients. Blood. 2011;117(10):2783-2790.
- Balagula Y, Straus DJ, Pulitzer MP, Lacouture ME. Necrobiotic xanthogranuloma associated with immunoglobulin m paraproteinemia in a patient with Waldenstrom macroglobulinemia. J Clin Oncol. 2011;29(11):e305-307.
- Cavalli G, Guglielmi B, Berti A, Campochiaro C, Sabbadini MG, Dagna L. The multifaceted clinical presentations and manifestations of Erdheim-Chester disease: comprehensive review of the literature and of 10 new cases. Ann Rheum Dis. 2013;72(10):1691-1695.
- Char DH, LeBoit PE, Ljung BM, Wara W. Radiation therapy for ocular necrobiotic xanthogranuloma. Arch Ophthalmol. 1987;105(2):174-175.
- Davies MJ, Whitehead K, Quagliotto G, Wood D, Patheja RS, Sullivan TJ. Adult Orbital and Adnexal Xanthogranulomatous Disease. Asia Pac J Ophthalmol (Phila). 2017;6(5):435-443.
- Ebrahimi KB, Miller NR, Sassani JW, Iliff NT, Green WR. Failure of radiation therapy in orbital xanthogranuloma. Ophthalmic Plast Reconstr Surg. 2010;26(4):259-264.
- Ghani S, Al Ustwani O, Khalid B, et al. Periorbital necrobiotic xanthogranuloma treated successfully with novel multiple myeloma therapy. Clin Adv Hematol Oncol. 2013;11(10):678-680.
- Green MB, Daly MK, Laver NMV, Lefebvre DR. Adult-onset asthma and periocular xanthogranuloma – A rare infiltrative disease of the orbit and eyelid. Am J Ophthalmol Case Rep. 2021;22:101043.
- Haroche J, Cohen-Aubart F, Emile JF, et al. Reproducible and sustained efficacy of targeted therapy with vemurafenib in patients with BRAF(V600E)-mutated Erdheim-Chester disease. J Clin Oncol. 2015;33(5):411-418.
- Hervier B, Haroche J, Arnaud L, et al. Association of both Langerhans cell histiocytosis and Erdheim-Chester disease linked to the BRAFV600E mutation. Blood. 2014;124(7):1119-1126.
- Kerstetter J, Wang J. Adult Orbital Xanthogranulomatous Disease: A Review with Emphasis on Etiology, Systemic Associations, Diagnostic Tools, and Treatment. Dermatol Clin. 2015;33(3):457-463.
- Liszewski W, Wisniewski JD, Safah H, Boh EE. Treatment of refractory necrobiotic xanthogranulomas with extracorporeal photopheresis and intravenous immunoglobulin. Dermatol Ther. 2014;27(5):268-271.
- Maeng MM, Godfrey KJ, Jalaj S, Kazim M. Adult xanthogranulomatous disease of the orbit: case report of spontaneous regression and review of treatment modalities. Orbit. 2020;39(1):31-37.
- McKelvie P, McNab AA, Hardy T, Rathi V. Comparative Study of Clinical, Pathological, Radiological, and Genetic Features of Patients With Adult Ocular Adnexal Xanthogranulomatous Disease, Erdheim-Chester Disease, and IgG4-Related Disease of the Orbit/Ocular Adnexa. Ophthalmic Plast Reconstr Surg. 2017;33(2):112-119.
- Miszkiel KA, Sohaib SA, Rose GE, Cree IA, Moseley IF. Radiological and clinicopathological features of orbital xanthogranuloma. Br J Ophthalmol. 2000;84(3):251-258.
- Nelson CA, Zhong CS, Hashemi DA, et al. A Multicenter Cross-Sectional Study and Systematic Review of Necrobiotic Xanthogranuloma With Proposed Diagnostic Criteria. JAMA Dermatol. 2020;156(3):270-279.
- Ortiz Salvador JM, Subiabre Ferrer D, Perez Ferriols A. Adult Xanthogranulomatous Disease of the Orbit: Clinical Presentations, Evaluation, and Management. Actas Dermosifiliogr. 2017;108(5):400-406.
- Papo M, Diamond EL, Cohen-Aubart F, et al. High prevalence of myeloid neoplasms in adults with non-Langerhans cell histiocytosis. Blood. 2017;130(8):1007-1013.
- Sahu K, Sethy M, Sirka CS, Rout AN. Adult-Onset Asthma with Periocular Xanthogranuloma (AAPOX), a Variant of Periorbital Xanthogranulomatous Disease: An Uncommon Entity. Indian Dermatol Online J. 2020;11(5):792-795.
- Satchi K, McNab AA, Godfrey T, Prince HM. Adult orbital xanthogranuloma successfully treated with rituximab. Ophthalmology. 2014;121(8):1664-1665 e1661-1663.
- Sivak-Callcott JA, Rootman J, Rasmussen SL, et al. Adult xanthogranulomatous disease of the orbit and ocular adnexa: new immunohistochemical findings and clinical review. Br J Ophthalmol. 2006;90(5):602-608.
- Spicknall KE, Mehregan DA. Necrobiotic xanthogranuloma. Int J Dermatol. 2009;48(1):1-10.
- Szalat R, Arnulf B, Karlin L, et al. Pathogenesis and treatment of xanthomatosis associated with monoclonal gammopathy. Blood. 2011;118(14):3777-3784.
- Toya T, Ogura M, Toyama K, et al. Prognostic factors of Erdheim-Chester disease: a nationwide survey in Japan. Haematologica. 2018;103(11):1815-1824
- Ugurlu S, Bartley GB, Gibson LE. Necrobiotic xanthogranuloma: long-term outcome of ocular and systemic involvement. Am J Ophthalmol. 2000;129(5):651-657.
- Zoumalan CI, Erb MH, Rao NA, et al. Periorbital xanthogranuloma after blepharoplasty. Br J Ophthalmol. 2007;91(8):1088-1089.